

 Anglais
Anglais français
français espagnol
espagnol russe
russe coréen
coréen japonais
japonaisÉtude sur différents Types d’extrait de Ginseng Ginsenoside
Ginsenosides (GS) are Le conseil des ministresmadansactifingredients dansprecious medicinal herbs such En tant queginseng, panax notoginseng Et en plusAmerican ginseng. They belong to Le conseil des ministrestriterpenoid glycoside class Et en plusare composed De laA AAAaglycoside (ginsenoside) Et en plusa sugar group [1]. Ginsenosides have a wide range De labiologiqueactivities, such as anti-tumeur[2], anti-inflammatoire[3], anti-fatigue [4-5] Et en plusanti-oxidatiSur le[6], etc., Et en plussecondary ginsenosides exhibit even more excellent activities. However, the highly active secondary ginsenosides have problems such as low content, pauvrewater solubility, low bioavailability and short half-life [7], which limits the applicatiSur leDe laginsenosides in the fields De lafood health care and biomedicine.
La modificatiSur lestructurelle est un moyen important d’améliorer l’activité biologique des ginsénosides, d’améliorer les propriétés pharmacocinétiques Et etde réduire la toxicité. Les ginsénosides sont généralement composés d’une aglycone hydrophobe liée à 1 à 4 groupements hydrophiles de sucre, de sorte qu’ils peuvent être modifiés de ces deux manières. Actuellement, la modification des ginsénosides adopte principalement des stratégies de modification chimique, Et etdes dérivés ginsénosides ayant une meilleure activité Et etdes propriétés physico-chimiques sont obtenus en modifiant la structure de l’aglycone ou en modifiant la chaîne du sucre [8]. En termes de structure, la structure cycloalkane de l’aglycone est stable, Et etil est difficile de modifier directement l’épine dorsale de l’aglycone.
Les principales stratégies actuelles de modification se concentrent sur le groupe hydroxyle sur l’aglycone, Et etdes dérivés de structures diverses sont obtenus par des méthodes synthétiques telles que l’estérification, l’oxydation, l’introduction d’hétérocycles, ou l’hybridation moléculaire. Les Modifications de la chaîne du sucre impliquent principalement l’extension de la fraction du sucre ou la modification du groupe hydroxyle sur la chaîne du sucre. Des études ont montré que le type, le nombre Et etle site de liaison du groupe sucre sur le noyau parent du ginsénoside sont étroitement liés à l’activité biologique des ginsénosides [18-20]. En général, la relation entre le nombre de groupes de sucre Et etl’activité anti-tumorale des ginsénosides est la suivante: aglycone > Glycoside monosaccharide > Disaccharide glycoside > Trisaccharide glycoside > Tétrasaccharide glycoside. Par conséquent, la modification des chaînes sucrières des ginsénosides est d’une grande importance pour améliorer leur activité biologique. CEt etarticle passe en revue les progrès récents dans la modification chimique Et etl’activité biologique des ginsénosides, énumère la relation structure-activité Et etrésume les caractéristiques Et etles lois de la modification structurale des ginsénosides, fournissant une référence pour les modifications structurales subséquentes.

1 Classification Et etcaractéristiques structurelles des ginsénosides
Selon les différentes structures de l’aglycone, ils peuvent être divisés en trois types: type dammarane, type oleanane Et ettype ocotillol. Les ginsénosides de type dammarane peuvent être subdivisés en protopanaxdiol (PPD) et propanaxatriol (PPT) en fonction de la position du groupe de substituant fixé à l’aglycone.
1.1 type de Dammarane
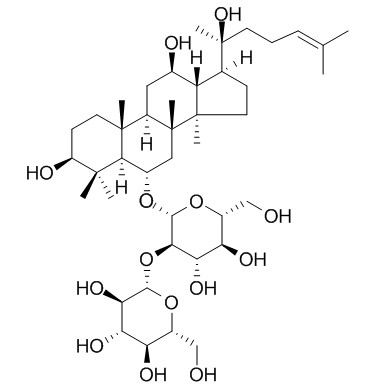
Dammarane-type ginsenosides include PPD and protopanaxatriol (PPT), which are tricyclic triterpene saponins. Common protopanaxadiol ginsenosides include C-K (1), L / 2(2), Rd (3), Rg3 (4), Rb1 (5), Ra1, Ra2, Ra3, Rb2 and panaxadiol (PD) (Fig. 1). Since ginsenosides 1, 2, 4 and 5 have stronger biological activity, leursynthèseand modification have attracted much attention [9-11]. Common protopanaxatriol-type ginsenosides mainly include Rh1 (6), Rg1 (7), Rg2 (8), Re, Rf, F1, F3, F5 and glycosylated panaxatriol (PT) (see Figure 2), among which 6, 7 and 8 have been studied extensively [8, 12].
1.2 type d’acide oléanolique
Les ginsénosides de type acide oléanolique sont des saponines triterpéniques pentacycliques, qui sont formées par la glycosylation de saponines de type oléanane (avoine) aux positions C-3 et C-28. Les ginsénosides communs de type acide oléanolique comprennent R3 (9), Ro (10) et R4 (11), entre autres [13-14] (voir Figure 3).
1.3 types d’ocotillol
Les Ocotillol typesapogénines (OTS) peuvent former quatre conformations: (20S,24S), (20R,24R), (20S, 24R) et (20R,24S), selon la configuration des groupes de sucre C-20 et C-24. Les ginsénosides courants du type Oxytropae comprennent F11 (12), RT5 (13), RT2 (14), etc. [15-17] (voir Figure 4).
2 modification structurelle des ginsénosides et relations structure-activité
2.1 modification d’estérification
Les ginsénosides ont une mauvaise solubilité dans l’eau et une faible solubilité dans les graisses, ce qui entraîne une faible biodisponibilité et un maintien de la santé et des effets thérapeutiques inefficaces. La lipophilicité idéale d’une molécule de médicament doit être dans une certaine plage pour assurer sa biodisponibilité et son efficacité clinique [8]. Des études pharmacocinétiques ont montré que les ginsénosides sont hydrolysés par la flore intestinale après administration orale, et que les métabolites produits par l’hydrolyse sont absorbés dans le foie par la veine, où ils réagissent avec les acides gras pour former des composés d’ester d’acides gras [21]. D’autres études ont révélé que les dérivés de ginsénoside liés aux acides gras ont une faible cytotoxicité dans les cellules, un long temps de séjour, et un effet plus durable. D’autre part, parce que les membranes cellulaires sont principalement composées de lipides, les dérivés d’ester lipophiles peuvent améliorer l’l’absorptionorale de médicaments indésirables en améliorant la perméabilité globale de la membrane. Ces résultats fournissent des idées pour la modification des ginsénosides. La modification des ginsénosides à l’aide d’acides tels que les acides organiques (acides gras, acides aromatiques, anhydrides), les acides aminés et les acides inorganiques (acide sulfurique) est une stratégie importante pour l’étude des dérivés ginsénosides.
2.1.1 modification des acides organiques
Liang et Al., et al.reacted the hydroxyl group at the C-3 position De laGinsénoside Rh2 (20S-Rh2, 2) with two 6-maleimidocaproic acid and 11-maleimidoundecanoic acid Produits dérivéswith hydrophilic functional groups and different carbon chain lengths to obtain the esterified Produits dérivés15 and 16 (see Figure 5). Compared with Rh2, the solubility De latwo modified products increased Par:about 4 times and 2 times, respectively. In vitro anti-proliferation Activité:tests showed that compound 15, which has a shorter carbon chain, exhibited higher inhibiteuractivity against the HeLa cell line, while compound 16, which has a longer carbon chain, did not show anti-proliferative activity [22]. Li et Al., et al.reacted decanoic acid, cyclohexanecarboxylic acid and isobutyric acid reacted with the C-20 hydroxyl group De laginsenoside C-K to synthesize three ginsenoside C-K monoesterified derivatives (17–19) [23] (see Figure 5).
En termes d’inhibition de la croissance de la lignée cellulaire du cancer du sein MCF-7, l’activité inhibiteur des composés 18 et 19 à 25 μmol/L était significativement plus élevée que celle du ginsénoside C-K,tandis que le composé 17 n’a montré aucun effet inhibiteur, ce qui indique que les dérivés du ginsénoside modifiés avec des acides gras à chaîne courte ont une activité anti-tumorale plus forte que ceux modifiés avec des acides gras à chaîne longue. D’autres études [24-27] ont également montré que modifiéshort-chain fatty acid saponin derivatives not only have optimized physicochemical properties, but also have better anti-tumor Les effetsthan long-chain fatty acid esters.
Li et Al., et al.ont synthétisé un dérivé entièrement acétylé du ginsénoside C-K par modification de polyestérification, à l’exception du groupe hydroxyle C-6 à base de glucose [28] (voir Figure 5). Les tests d’activité anti-tumorale ont montré que par rapport au C-K,il peut inhiber la prolifération de plusieurs lignée de cellules tumorales à des concentrations plus faibles, tout en inhibant significativement la croissance tumorale dans un modèle de xénogreffe de carcinome hépatocellulaire sans effets secondaires sur les principaux organes.
On peut constater qu’après estérification modification du ginsénoside C-K, sa cytotoxicité est réduite et son activité antitumorale est augmentée. Wang et Al., et al.ont réformé le groupe hydroxyle C-3 de la PD avec des dérivés d’acide benzoïque, des acides aminés et de l’anhydride tétrachlorophtalque pour obtenir une série de dérivés de la PD 21-31 [29] (voir Figure 5). Les tests de prolifération anti-tumorale ont montré que la plupart des composés avaient des effets inhibiteurs sur les lignées de cellules cancéreuses, y compris les cellules cancéreuses du foie humaines hépatig-2, les cellules cancéreuses du poumon humaines A549, les cellules cancéreuses du sein humaines MCF-7 et les cellules cancéreuses du côlon humaines HCT-116. Par rapport à la PD,les dérivés de ginsénoside 22, 23 et 26 ont montré des effets inhibiteurs significatifs sur la prolifération des cellules cancéreuses. Par exemple, 22 présentait la ci50 la plus faible pour l’a549 (ci50 = 18,91 ± 1,03 μmol/L), tandis que pour les cellules MCF-7, le composé 23 présentait une meilleure activité inhibitrices (ci50 = 8,62 ± 0,23 μmol/L). Ce résultat montre que l’introduction d’un acide aromatique dans les ginsénosides peut également améliorer significativement l’activité antitumorale.
La relation structure-activité mentionnée ci-dessus montre que l’introduction d’acides gras à chaîne courte dans les ginsénosides présente une meilleure activité que les acides gras à chaîne longue. Le nombre de sites de modification d’estérification (monoesters et polyesters) et le type d’acide (acides gras et acides aromatiques) n’ont pas d’effet significatif sur l’activité biologique.
2.1.2 modification des acides aminés
Le 25-Hydroxyl-protopanaxdiol (25-OH-PPD) (34), un composé naturel isolé des fruits de ginseng, a une activité antitumorale significative et les avantages de faibles effets secondaires et une biodisponibilité orale absolue élevée. Yuan et Al., et al.[30] l’ont combiné avec des acides aminés non protéiques qui jouent des fonctions physiologiques uniques et des valeurs médicinales, ont conçu et synthétisé une série de nouveaux dérivés 25-OH-PPD 33-45 (voir Figure 6). L’activité antitumorales a montré que certains dérivés 25-OH-PPD ont montré une excellente activité inhibiteur contre la prolifération tumorales. Par exemple, le composé 33 a montré une forte activité antitumorale contre les lignées cellulaires HCT116 et BGC-823, avec des valeurs de ci50 de 4,76 μmol/L et 6,38 μmol/L,respectivement (voir tableau 1). De plus, des dérivés d’acides aminés de 25-OH-PPD (46-59) ont également montré une activité antitumorale [31] (voir tableau 1).
Comme le montre le tableau 1, les valeurs de ci50 de certains produits modifiés par des acides aminés non protéiques sont supérieures à 100 μmol/L, tandis que l’activité anti-tumorale des produits modifiés par des acides aminés protéiques est généralement meilleure que celle des produits modifiés par des acides aminés non protéiques, et leurs valeurs de ci50 sont toutes inférieures à 30 μmol/L. D’autre part, les acides aminés sur les dérivés de ginsénoside ayant une valeur ci50 inférieure à 10 μmol/L pour la prolifération anti-tumorale ont tous des groupes protecteurs de la Boc, et l’élimination des groupes protecteurs de la Boc réduit de manière significative l’activité anti-tumorale du produit, ce qui indique indirectement que l’augmentation de la solubilité lipidique du produit par estérification peut augmenter de manière significative l’activité biologique des dérivés de ginsénoside.
2.1.3 modification des acides inorganiques
At present, inorganic acid modification mainly uses the sulfonating reagentchlorosulfonic acid to react with the hydroxyl groups on the ginsenoside sugar chain to form a sulfonate, which is then converted to a salt by neutralization with pyridine. As the introduction De laa sulfate group increases the polarity De laginsenoside derivative, solubility is improved. It has been reported that the anticancer activity of sea cucumber saponins, which have a similar structure to ginsenosides, is related to the sulfate group,the fewer sulfate groups present on the sugar chain, the stronger the Lutte contre le canceractivity [32].

Based on these findings, Guo et Al., et al.[33] used the chlorosulfonic acid-pyridine method to sulfate modify ginsenosides. The resulting derivative SMTG-d3 enhanced naturelkiller cell activity by promoting the proliferation of T lymphocytes and the production of IFN-γ and TNF-α cytokines. Compared with ginsenoside, SMTG-d3 not only reduces cytotoxicity, but also further enhances antitumor immune activity. Previously, Fu et Al., et al.[34-36] also used this method to convert the C-6 hydroxyl group De 20(S)-ginsenosideL / 2to a sulfonate ester and synthétisétwo new derivatives 60 and 61 with greatly improved solubility (see Figure 7). Further studies have found that both derivatives can enhance anti-inflammatoireand immune Les effetsby blocking mitogen-activated protein kinase and the Communiqué de presseof pro-inflammatory mediators induitby activation. This shows that the sulfation of ginsenoside derivatives can increase their solubility, thereby enhancing Activités activitéssuch as anti-inflammatory and anti-tumor effects.
2.2 modification oxydative
La double liaison planaire sur la chaîne latérale ginsénoside et le groupe hydroxyle dans la structure aglycone fournissent des sites de réaction pour la modification de l’oxydation, permettant d’oxyder la chaîne latérale C-17 et les anneaux A et C de certains ginsénosides. Des études ont montré que la double liaison sur la chaîne du côté ginsénoside est l’une des raisons de sa faible solubilité [37]. Les ginsénosides peuvent augmenter leur solubilité dans l’eau en réduisant le degré d’insaturation ou en ajoutant des groupes ionisables tels que les groupes carboxyle par modification oxydative, améliorant ainsi leur activité biologique.
Wong et Al., et al.[38] ont oxydé la double liaison sur la chaîne latérale du ginsénoside 20(R)-Rh2 (2) pour obtenir un dérivé 20(R)-Rh2E2 (62) qui peut efficacement prévenir le développement du cancer colorectal induit par l’azométhane oxydé/sel sulfate de sodium de dextran (AOM/DSS) (voir Figure 8). Ce composé époxyde a également une activité inhibitoire contre d’autres lignée de cellules cancéreuses. Par exemple, sa ci50 pour les cellules cancéreuses du poumon (LLC-1) est de 56 μmol/L.
Il a été constaté que la PPD est métabolisée dans le foie humain pour former de l’époxyde C-20-24, qui contient le squelette Pyxinol eta une bonne activité anti-inflammatoire [39]. Wang et Al., et al.ont époxydé 20(S)-PPD, puis ont subi une oxydation Dess-Martin, une réduction sélective avec du NaBH4, des réactions de condensation et de déprotection pour obtenir une série de dérivés de Pyxinol oxydés C-12 modifiés par des acides aminés [40] (voir la Figure 9).
Using the Griess method to test the inhibitory activity of these derivatives on nitric oxide in RAW264.7 macrophages, derivatives 63a, 63b, 63c, 63d, 64e, 66b, and 66c showed good anti-inflammatory activity (inhibition rates of 48% to 85%), even better than Y13 (known as the Pyxinol derivative with the best anti-inflammatory activity at the C-12 site, with a hydroxyl group, and an inhibition rate of <40%) and the clinically approved glucocorticoid steroid drug hydrocortisone sodium succinate. A structure-activity relationship study showed that oxidation of Pyxinol at the C-12 position can effectively improve the anti-inflammatory activity of derivatives modified at the C-3 position. In particular, N-Boc-protected aromatic amino acids can significantly enhance their anti-inflammatory activity. At the same time, derivatives with the absolute configuration of R at the C-24 position are more active.
Wang et Al., et al.[41] ont utilisé une méthode similaire pour oxyder sélectivement la position C-3 de l’anneau du squelette a du Pyxinol et introduire simultanément un accepteur Michael pour préparer 24 nouveaux dérivés du ginsénoside (67a-67h, 68a-68h, 69a-69h) (voir la Figure 10). La relation structure-activité montre que la fusion de ginsénoside PPD avec un accepteur Michael peut améliorer l’activité anti-inflammatoire du dérivant, et la présence d’un groupe de retrait d’électron sur l’accepteur Michael améliore encore l’activité anti-inflammatoire. L’activité anti-inflammatoire du dérivé obtenu en modifiant la position C-20 de PPD avec un anneau tétrahydrofuran a été considérablement réduite, mais l’activité anti-inflammatoire du dérivé dans lequel l’anneau a a été oxydé a été presque inchangée, ce qui indique en outre que l’activité biologique anti-inflammatoire de certains ginsénosides peut être considérablement améliorée par la modification de l’oxydation.
Zhang et Al., et al.[42] ont hydrolysé le PD et l’ont oxydé à l’aide du pyridinechlorochromate (PCC), de l’o2 et du H2O2 pour obtenir une série de chaînes latérales C-17 et de dérivés d’oxydation des anneaux a et C (voir la Figure 11). Les tests de cellules antitumorales ont montré que certains composés présentaient une meilleure activité antiproliférative que le contrôle positif dans six lignées cellulaires, y compris A549 (cancer du poumon humain), 8901 (cancer de l’ovaire humain), et d’autres lignées cellulaires. Par exemple, dans la lignée cellulaire U87 (gliome humain), les composés 70, 78, 82 et 83 se sont révélés plus efficaces que le témoin positif, le composé 82 ayant une ci50 de 19,51 ± 1,00 μmol/L. Dans la lignée cellulaire MCF-7 (cancer du sein humain), comparativement au 5-fluorouracile et au PD, les composés 71 et 82 présentaient une meilleure activité antitumorale (ci50 = 17,73 ~ 23,58 μmol/L); Les composés 71 et 74 ont également montré une bonne activité antiproliférative dans les cellules HeLa. Des études ont montré que l’introduction d’une structure énol au site α de l’anneau A des dérivés PD peut améliorer leur activité antitumorale, mais pas toutes les modifications oxydatives des dérivés PD atteindre cet effet. Par exemple, l’activité antiproliférative du composé 81, obtenue par oxydation ultérieure avec H2O2, a été réduite.
2.3 modification hétérocyclique
Les composés hétérocycliques sont souvent utilisés dans la conception et la synthèse de médicaments en raison de leur diversité structurelle et de leur large éventail d’activités biologiques, ce qui permet d’élargir l’espace disponible pour la chimie de type médicament. La plupart des médicaments commercialisés contiennent des structures hétérocycliques, les hétérocycles d’azote étant les plus courants dans les structures de médicaments commercialisés. Les hétérocycles d’azote et les hétérocycles d’oxygène couramment présents dans les molécules médicamenuses contiennent des paires d’électrons isolés, qui peuvent former des liaisons d’hydrogène, ce qui favorise l’amélioration de la solubilité dans l’eau et donc de la biodisponibilité. Les anneaux de pipérazine et les anneaux de pipéridine sont des structures hétérocycliques d’azote très courantes dans les médicaments commercialisés. Elles peuvent être dérivées davantage pour établir une petite bibliothèque de composés [43], ce qui favorise la conception d’un plus grand nombre de composés pour des études approfondies de la relation structure-activité. Des études ont montré que l’introduction d’hétérocycles dans les produits naturels peut considérablement améliorer l’activité biologique et la solubilité des dérivés par le principe de la "combinaison pharmacophore" et une augmentation du nombre de liaisons d’hydrogène [44-45]. A l’heure actuelle, un grand nombre d’études ont fait état de la modification des dérivés ginsénosides avec les hétérocycles. Parmi eux, les composés hétérocycliques contenant de l’azote ont une faible cytotoxicité et présentent une bonne solubilité dans l’eau, perméabilité et biodisponibilité.
Les Pyrazoles sont des composés hétérocycliques à cinq membres composés de deux atomes d’azote adjacents. Ils ont une variété d’activités pharmacologiques telles que des effets anti-inflammatoires, antiviraux et antidépresseurs, et sont largement utilisés dans le développement de nouveaux médicaments [46]. Les dérivés d’isoxazole ont également une variété d’activités biologiques telles que des effets antibactériens, antiviraux et antitumoraux, et sont largement utilisés dans la synthèse organique [47]. Sur cette base, Dai et Al., et al.[48] ont introduit les squelettes pyrazole et isoxazole dans la position C-3 de PD, ont conçu et synthétisé 19 dérivés de PD contenant des hétérocycles (voir Figure 12), et ont étudié leur activité antiproliférative contre quatre cellules tumorales différentes. Les résultats ont montré que les produits 86 et 87 obtenus par fusion de l’anneau A de PD avec l’anneau pyrazole ont une activité anticancéreuse significative. Par exemple, 86 a une ci50 de 14,15 ± 1,13 μmol/L contre les cellules HepG-2, et 87 a une ci50 de 13,44 ± 1,23 μmol/L contre A549, soit quatre fois celle de PD. Il a également un plus grand effet inhibiteur sur les trois autres cellules tumorales. Cependant, les composés 88 et 89a-89i présentent une faible solubilité dans l’eau en raison de la présence de multiples liaisons ester et de groupes hydrophobes tels que les anneaux aromatiques, ce qui entraîne une faible activité de prolifération anti-tumorale. La substitution des liaisons ester dans les structures des dérivés 88 et 89a-89i par des liaisons amide pour obtenir des dérivés 90a-90f n’a pas augmenté l’activité de prolifération anti-tumorale. D’autre part, les résultats des tests d’activité in vitro ont montré que 87 > 86,89a et gt; 88 et 90a et gt; 91, ce qui indique que les dérivés de PD modifiés par le pyrazole sont généralement plus actifs que les composés d’isoxazole.
Les composés Pyrazine et pyrimidine présentent un large éventail d’activités biologiques, et ces deux types de structures sont souvent trouvés dans les molécules médicamenuses commercialisées [49-50]. Wang et Al., et al.[51] ont introduit des hétérocycles tels que la pyrazine, l’oxadiazole, l’isoxazole, le pyrazole et la pyrimidine les hétérocycles ont été introduits dans les positions C-2 et C-3 de la PPD par des réactions organiques classiques telles que l’oxydation, l’hydrogénation, la condensation de l’ester de Claisen, la réduction et la protection et la déprotection des groupes hydroxyle. Une série de dérivés hétérocycliques de 20(S)-PPD fusionnés A été synthétisée (voir Figure 13) et leurs effets inhibiteurs sur l’activateur de récepteur de la différenciation ostéoclaste induite par le ligand κ B (RANKL) du facteur nucléaire ont été évalués. La relation structure-activité montre que par rapport à la PPD, en plus des dérivés du phénylpyrazole, l’inhibition de la différenciation de l’ostéoclast par les dérivés oxadiazole, isoxazole et pyrazole avec des modifications hétérocycliques à cinq cycles (93, 94 et 95a) ont une activité inhibiteure similaire ou légèrement plus forte (ci50 = 10,3 μmol/L) que la PPD; Alors que l’activité inhibiteure de composés modifiés avec des cycles hétérocycliques à six cycles tels que la pyrazine et la pyrimidine (92, 96a) est considérablement augmentée.
Sur la base de l’excellente activité du composé 96a, le groupe de recherche a encore modifié les dérivés de la pyrimidine (voir Figure 14). Les résultats ont montré qu’à une concentration modérée de 1,0 μmol/L, la plupart des dérivés avaient un effet inhibiteur de presque 100% (sauf 96f); À une concentration de 0,1 μmol/L, l’effet d’inhibition des dérivés modifiés méthyle (96b) et éthyle (96c) a été considérablement accru, tandis que l’activité d’inhibition des composés modifiés méthoxy (96d), éthoxy (96e) et aminé (96g) est demeurée presque inchangée. Les chercheurs ont ensuite modifié la structure 96b avec une chaîne latérale C-12-hydroxy ou C-17 (voir Figure 14). Les résultats ont montré que le remplacement du groupe hydroxyle en position C-12 par un groupe cétone (98), oxime (99), α-hydroxy (100) ou acétate (101) a entraîné une diminution significative de l’activité inhibiteur. À une concentration de 0,01 μmol/L, le 98-101 ne présentait presque aucun effet inhibiteur. Le composé 105 présentait la meilleure activité inhibiteur (ci50 = 11,8 nmol/L), même à une concentration de 0,01 μmol/L, ce qui était meilleur que l’activité PPD (ci50 = 10,3 μmol/L), et il pouvait inhiber l’ostéoclastogenèse à la fois in vitro et in vivo.
2.4 modification des polymères
Les médicaments anticancéreux modifiés par polymères hydrophiles peuvent non seulement compenser leur faible ciblage, mais aussi améliorer la solubilité dans l’eau, la stabilité, la demi-vie in En directet la biodisponibilité du médicament [52]. Ces dernières années, la technologie d’administration des médicaments s’est développée rapidement, ce qui rend les ginsénosides largement étudiés. Lu et al. [53] ont préparé des conjugués du Rh2 avec du o-carboxyméthyl chitosan (O-CMC) soluble dans l’eau (O-CMC conjugué au Rh2, O-CMC/Rh2) (106) (voir la Figure 15) au moyen d’une réaction d’estérification. Les résultats ont montré que 106 était très poreux et que les liaisons ester dans la structure étaient sensibles au pH. À pH H5,8, le taux de libération de Rh2 était plus rapide au stade précoce, de sorte que le taux de libération de Rh2 pouvait être contrôlé en fonction des changements de pH au site de la blessure pendant la douleur inflammatoire. Le conjugué de o-carboxyméthyl chitosan a amélioré l’efficacité biologique du Rh2 in vivo en augmentant sa solubilité, en régulant son taux de libération, et en prolongeant sa durée d’action dans le corps. Améliorant ainsi l’efficacité biologique du Rh2 in vivo.
Le polyéthylène glycol (PEG) présente les avantages d’être facile à modifier, biodégradable, biocompatible et d’avoir un taux élevé d’encapsulation des médicaments, et se montre donc très prometteur dans l’administration des médicaments. Mathiyalagan et al. [54] ont combiné le PEG hydrophile avec le Rh1 et le Rh2 hydrophobes pour synthétiser deux types de dérivés de ginsénoside à administration passive ciblée (voir la Figure 15). Comparativement au Rh1, le PEG-Rh1 (107) a une activité antitumorale plus élevée dans les lignées cellulaires humaines cancéreuses du poumon (A549), tandis que le PEG-Rh1 et le PEG-Rh2 ne présentent pas de cytotoxicité chez une lignée cellulaire de macrophages murins non infectés (crue 264.7). Parmi eux, le PEG-Rh2 (108) peut considérablement inhiber la production d’oxyde nitrique et ainsi présenter une meilleure activité anti-inflammatoire. Cela indique que les polymères PEG peuvent non seulement améliorer la solubilité des ginsénosides et réduire la cytotoxicité, mais aussi atteindre une administration ciblée grâce à l’effet amélioré de perméabilité et de rétention (EPR) et à différentes conditions de pH.
2.5 modification conjuguée
Il a été rapporté que les conjugués TPP ont une forte capacité de ciblage mitochondrial, et ont été utilisés pour administrer sélectivement des médicaments anticancéreux, y compris l’adriamycine et le cisplatine, aux mitochondries des cellules tumorales [55]. Afin d’améliorer le ciblage et l’activité du ginsénoside 25-MeO-PPD, 25-OH-PPD et PD, Ma et al. [56] ont introduit des chaînes alkylées de différentes longueurs à leur position C-3, puis de la triphénylphosphine conjuguée (TPP) à la fin pour synthétiser une série de conjugués du ginsénoside (voir la Figure 16). Des études anti-prolifération sur les lignées de cellules cancéreuses (A549, MCF-7) et les cellules normales (GES-1) ont montré que la plupart des conjugués étaient plus actifs que les composés parentaux correspondants, et ont montré des effets inhibiteurs plus forts dans les cellules cancéreuses que dans les cellules normales. Parmi eux, 109 peuvent s’accumuler dans les mitochondries des cellules MCF-7, stimuler la production d’espèces réactives d’oxygène (ROS), et provoquer une dépolarisation du potentiel de la membrane mitochondriale, conduisant à l’apoptose. Par conséquent, 109 présente une sélectivité élevée et un bon effet antiprolifératif (ci50 = 0,76 μmol/L) sur les cellules MCF-7.
2.6 autres modifications
En plus des cinq méthodes mentionnées ci-dessus, les modifications structurelles des ginsénosides comprennent également l’éthérification, l’alkylation, l’hydrogénation catalytique et la glycosylation [57-58]. L’éthérification et l’alkylation impliquent la réaction du groupe hydroxyle des ginsénosides avec les haloalcanes sous la catalyse des alcalis. L’hydrogénation catalytique implique l’utilisation d’un catalyseur pour hydrogéner directement le groupe insaturé dans la structure monomère des ginsénosides en un groupe saturé. La Glycosylation implique l’introduction de groupes donneurs correspondants tels que les groupes mannosyl, xylosyl et rhamnosyl dans le groupe hydroxyle des ginsénosides. Par exemple, Ren et al. [59] ont introduit des donneurs de sucre au C-20 OH des dérivés de PPD par oxydation, réduction, substitution nucléophile, et d’autres réactions pour préparer une série de dérivés de ginsénoside C-K avec différents anneaux de sucre [60-65].
3 résumé
Cet article passe en revue les méthodes de modification structurelle des ginsénosides au cours des dernières années. Il utilise principalement des acides organiques, des acides aminés et des acides inorganiques pour réagir avec les groupes hydroxyle aux positions C-3 et C-20 de l’aglycone et les groupes hydroxyle primaires sur la chaîne du sucre des ginsénosides pour obtenir des dérivés ester, afin d’améliorer la solubilité lipidique et la biodisponibilité des ginsénosides. Des études sur les relations structure-activité ont montré que l’activité des dérivés de ginsénoside après modification d’estérification a les caractéristiques suivantes: Acides gras saturés, acides gras à chaîne courte et gt; Acides gras à longue chaîne. La modification des sulfates en introduisant des groupes polaires, la modification de l’oxydation en réduisant l’insaturation ou en ajoutant des groupes ionisables tels que les groupes carboxyliques, la modification hétérocycliques en augmentant le nombre de liaisons d’hydrogène et la modification des complexes hydrophiles peuvent toutes améliorer la solubilité dans l’eau et la biodisponibilité des ginsénosides à des degrés divers, et peuvent considérablement améliorer l’activité biologique de ces dérivés. Ces méthodes de modification constituent une référence importante pour l’étude, le développement et l’application des ginsénosides.
However, there are currently some deficiencies in the structural modification of ginsenosides: firstly, there are relatively few structural modification methods. Structural modification mainly involves introducing groups that can react with hydroxyl groups, making the sites and types of products of structural modification relatively simple, and leading to insufficient research on the structure-activity relationship. In particular, there is a relative lack of modification of the sugar chain. There are few reports on the replacement of the sugar chain and the splicing of the sugar chain essential for activity with other different types of aglycon or skeleton to enhance activity and broaden the scope of activity. Second, the lack of precision in structural modification results in low activity. Current research on the activity of ginsenoside derivatives mainly focuses on in vitro anti-tumor and anti-oxidant activities, and there is relatively little further research on in vivo activity, with very few compounds entering clinical research. Third, the research on the activity of ginsenoside derivatives is not in-depth enough. There is very limited research on ginsenosides and their derivatives with immunostimulatory activity as vaccine adjuvants. The few studies on ginsenoside adjuvants mainly use crude ginsenoside extracts, and there is a lack of systematic research on the potential application value of ginsenosides and their derivatives as potent immunostimulants in vaccine adjuvants.
Quatrièmement, les modifications structurelles actuelles se concentrent principalement sur les ginsénosides de type dammarane, avec relativement peu de modifications pour l’acide oléanolique et les ginsénosides de type orcinélo. Par conséquent, les modifications structurelles futures devraient améliorer la précision de l’introduction des groupes et des structures, élargir les méthodes de modification structurelle et les sites de modification, élargir le champ d’application des dérivés de ginsénoside, et jeter une base théorique pour le développement des médicaments ginsénoside et des aliments de santé.
Référence:
[1]HOU M, WANG WANGWANGR, ZHAO S, et al. Ginsénosides du genre panax et leur biosynthèse [J].Acta Pharmaceutica Sinica B,2021, 11 (7): 1813-1183.
[2]WANG Y YYH,AI Z ZY,ZHANG ZHANGZHANGZHANGJ D, et al. Progrès de la recherche sur l’activité antitumorale et le mécanisme des ginsénosides [J].Science and Technology of Food Industry, 2023, 44(1): 485-491.
[3]PAIk S, SONG G Y,JO E K, et al. Ginsénosides pour cibler l’inflammation par modulation du stress oxydatif [J].InternationalImmunopharmacology, 2023, 121: 110461.
[4]LIU LIUF X,LIN Z X,ZHANG H L, et al. Analyse du mécanisme anti-fatigue et des cibles potentielles du ginseng [J].China Revue de presseof Chinese Materia Medica, 2019, 44(24): 5479-5487.
[5]ARRING N M, MILLSTINE D, MARKL lL A, et al. Le Ginseng comme traitement de la fatigue: une revue systématique [J]. JAltern complément Med. 2018, 24(7):624-633.
[6]FENG S, LI T, T,WEI X, ZHENG ZHENGY,et al. Les effets antioxydants et anti-Fatigue des ginsénosides rares et de l’acide γ-aminobutyrique dans le ginseng fermenté et la purée de riz brun germée. International Revue de presseof Molecular Sciences. 2024, 25(19):10359.
[7]HU Q QQ R, HONG kong H, ZHANG Z H, et al. Méthodes de travail on Améliorations apportées of the poor Par voie orale Biodisponibilité des ginsénosides: pré-traitement, modification structurelle, combinaison de médicaments et système de micro- ou nano- administration [J]. Journal of Ginseng Research, 2023, 47 (6): 694-705.
[8]FunL, l,FAN L, WANG Z,et al. Ginsénosides rares: une perspective unique de la recherche sur le ginseng [J]. Journal of Advanced Research, 2024, https://doi.org/10.1016/j.jare.2024.01.003.
[9]XU L, l,LYU W, DUAN C,et al. Préparation et bioactivité des rares ginsénosides Rg3 Et Rh2: une revue mise à jour [J]. Fitoterapia, 2023, 167: 105514.
[10]LI S, LI J, J,J,J,J,ZHAO Y,et al. Intégration supramoléculaire de nanomatériau multifonctionnel par azocalixarène décoré de mannose avec ginsenoside Rb1 Pour la thérapie synergique de la polyarthrite rhumatoïde [J]. ACS Nano, 2023, 17 (24): 25468-25482.
[11]YAN H,JIN H, FU Y,et al. Panax ginseng production de ginsénosides rares Rg3 Et Rh2 par des bactéries endophytes de [J]. Journal of Agriculture and Food Chemistry, 2019, 67 (31): 8493-8499.
[12]XU X, QU W, JIA Z, et al. Effet des âges de culture sur l’activité anti-inflammatoire d’un nouveau type de ginseng rouge [J]. Biomédecine &; Pharmacotherapy, 2021, 136: 111280.
[13]BEDNARCZYK-CWYNAR B,le
[14]DENG X, KE J, ZHENG Y,et al. α synthesis Évaluation des projetsdes bioactivités de l’oléanolique Dérivés de l’acide oxime ester en tant qu’inhibiteurs de -glucosidase et -amylase [J]. Journal of Enzyme Inhibition and Medicinal Chemistry, 2022, 37 (1): 451-461.
[15]CAO CAOY, WANG K, WANG J, et al. Conception, synthèse et évaluation antibactérienne de dérivés d’ocotillol avec des groupes polycycliques contenant de l’azote [J]. Future Medicinal Chemistry, 2021, 13 (12): 1025-1039.
[16]LIU J, GAN H, LI T et al. Les métabolites et les voies de La biotransformationin vivo après administration orale d’ocotillol [J]. Biomedical Chromatography, 2020, 34 (8): e4856.
[17]ZHANG D, CAO Y, WANG K, et al. La conception, Synthèse, and antibactérien Évaluation des projets De roman Dérivés d’ocotillol et leurs effets synergiques avec les antibiotiques conventionnels [J]. Molécules, 2021, 26 (19): 5969.
[18]TONG Y, SONG X, ZHANG Y, et al. Aperçu sur la modification structurelle, l’activité biologique, la relation structure-activité des dérivés ginsénoside de type PPP [J]. Fitoterapia, 2022, 158: 105135.
[19]GUO H, XING Y, Le soleil Y, et al. ginsengenine derivatives synthesized À partir de 20(R)-panaxotriol: La synthèse, la caractérisation et l’activité antitumorale ciblant la voie HIF-1 [J]. Journal of Ginseng Research, 2022, 46 (6): 738-749.
[20]HU Q, HONG H, ZHANG Z, et al. L’invention concerne des procédés d’amélioration de la faible biodisponibilité orale des ginsénosides: pré-traitement, modification structurelle, combinaison de médicaments et système de micro- ou nano- administration [J]. Journal of Ginseng Research, 2023, 47 (6): 694-705.
[21]LI J, DAI DAI Y L, ZHENG F, et al. Par voie orale absorption and in vivo biotransformation De ginsénosides [J]. Chinese Journal of Biologicals, 2014, 27(12):1633-1636.
[22]LIANG J, TANG X, WAN S, et al. Modification de la Structure du ginsénoside Rh2 Et l’activité cytostatique sur les cellules cancéreuses [J]. ACS Omega, 2023, 8 (19): 17245-17253.
[23]LI K K, YAN X M, LI Z N, et al. Synthèse et activité antitumorale de trois nouveaux ginsenoside M1 Dérivés avec modifications 3’-ester [J]. Bioorganic Chemistry, 2019, 90: 103601.
[24]HUANG Y, LI H M, ZHANG Y X, et al. La synthèse et l’évaluation biologique des dérivés K du composé ginsénoside en tant que nouvelle classe d’activateur lxrα [J]. Molécules, 2017, 22 (7): 1232.
[25]WANG R, LI M, LIU M, et al. Caractérisation de l’émulsion de pickering par de l’amidon déramifié modifié par scfa et a Puissant pour délivrer encapsulé Composé bioactif [J]. International Journal of Macromolécules biologiques, 2023, 231: 123164.
[26]SUNG KEE R, TIMONTHY J K, KAZUTOSHI F, et al. Effet d’un promédicament oral d’astaxanthine (CDX-085) sur les taux de lipoprotéines et la progression de l’athérosclérose chez les souris LDLR -/- et apoe -/- [J]. Athérosclérose, 2012, 222.
[27]CHRISTOPHER T C, UDAYANATH A, CHRISTOPHER A W, et al. Le ciblage d’oncogènes pro-invasifs avec des analogues d’acide gras à chaîne courte et d’hexosamine inhibe la mobilité des cellules métastatiques du cancer du sein mda-mb-231 [J]. Journal de chimie médicinale, 2008, 51:8135-8147.
[28]ZHANG J, TONG Y, LU X et al. L’invention concerne un dérivé du ginsénoside C-K et son effet inhibiteur sur le carcinome hépatocellulaire [J]. Life Sciences, 2022, 304: 120698.
[29]XIAO S, LIN Z, WANG X et al. Synthèse et évaluation de la cytotoxicité des dérivés du panaxadiol [J]. Chimie &; Biodiversité, 2020, 17 (1): e1900516.
[30]YUAN W,GUO J, WANG X,et al.dérivés non protéiques d’acides aminés du 25-méthoxylprotopanaxadiol /25-hydroxyprotopanaxadiol et leur anti-tumor activity Évaluation des projets [J]. Stéroïdes, 2018, 129: 1-8.
[31]LIN L, ZHAO Y, WANG P et al. Les dérivés d’acides aminés du ginsénoside AD-2 induisent l’apoptose des cellules HepG2 en affectant le cytosquelette [J]. Molécules, 2023, 28 (21): 7400.
[32]MIYAMOTO T, TOGAWA K, HIGUCHI R, et al. Six Nouvellement créé identifié biologiquement active Sulfates de glycoside triterpénoïde du concombre de mer cucurnaria echinata [J]. Liebigs Annalen der Chemie, 1989 1990 (5):453-460.
[33]GUO Z, WANG L, HAQ S, et al. Évaluation in vitro de l’activité immunomodulatrice du dérivé total de ginsénosides modifié par sulfatation 3 [J]. Frontières en sciences vétérinaires, 2023, 10: 1068315.
[34] ce que nous avons fait B, BI BIBI W, Il a C, et al. sulfatées derivatives of 20(S)-ginsenoside Rh2 and their inhibitory effects Sur les cytokines inflammatoires induites par les LPS et les médiateurs [J]. Fitoterapia, 2013, 84: 303-307.
[35]BI W, FU B, SHEN enH et al. Dérivé sulfaté de 20(S) -ginsénoside Rh2 Inhibe les cytokines inflammatoires par les voies mapks et nf-kappa b dans les macrophages RAW264.7 induits par LPS [J]. Inflammation, 2012, 35: 1659-1668.
[36]YIP, BI W, SHEN H, et al. inhibiteur effects of sulfatées 20(S)-ginsenoside Rh2 on the release of Médiateurs pro-inflammatoires dans des cellules brutes 264.7 induites par LPS [J]. European Journal of Pharmacology, 2013, 712: 60-66.
[37]ZHOU W X, YANG N, ZHAO Y Q. progrès dans les études sur l’amélioration de la solubilité dans l’eau du ginsénoside [J]. Drug Evaluation Research, 2016, 39(2): 322-327.
[38]WONG V, DONG H, LIANG X, et al. Le Rh2E2, un nouveau suppresseur métabolique, inhibe spécifiquement le métabolisme énergétique des cellules tumorales [J]. OncoTargets and Therapy, 2016, 7 (9): 9907-9924.
[39]SUN Y X, FANG X J, GAO N ° de catalogueet al. La présente invention concerne la synthèse et la relation structure - activité de dérivés de pyxinol en tant que nouveaux Les agentsanti-inflammatoires [J]. Lettre ACS sur la chimie médicinale, 2020, 11, 457-463.
[40]WANG Y, mon X, DU Y, et al. La conception, Synthèse, and anti-inflammatory activities of Dérivés du 12-déhydropyxinol [J]. Molécules, 2023, 28 (3): 1307.
[41]YANG G, Mi X, WANG Y et al. La Fusion des michael-accepteurs améliore l’activité anti-inflammatoire des ginsénosides en tant que modulateurs potentiels de la NLRP3 Voie de signalisation [J]. Chimie bioorganique, 2023, 134:106467.
[42]ZHANG Y M, YUAN W H, WANG X D, et al. Synthèse, caractérisation et évaluation de l’activité cytotoxique de l’oxydation du ginsengdiol et des dérivés hybrides d’azote [J]. MedChemComm, 2018, 9(11): 1910-1919.
[43]XIAO S, WANG X, XU L, et al. De nouveaux dérivés de ginsénoside ont montré leurs effets sur les cellules PC-3 en induisant l’arrêt de la phase g1 et l’apoptose réactive des cellules intermédiaires de l’espèce d’oxygène [J]. Bioorganic Chemistry, 2021, 112: 104864.
[44]YANG G Q, LIU S, ZHANG C et al. Découverte de dérivés pyxinol amide contenant des résidus d’acides aminés en tant que non-substrat allostérique Les inhibiteurs De la p-glycoprotéine médiée multidrogue résistance [J]. Journal of Medicinal Chemistry, 2023, 66 (13): 8628-8642.
[45]MA L, MIAO D, LEE J et al. Synthèse et évaluation biologique de dérivés de ginsénoside de type dammarane hétérocycliques fusionnés en anneaux en tant qu’agents antitumoraux potentiels [J]. Bioorganic Chemistry, 2021, 116: 105365.
[46]KARROUCHI K, RADI S, RAMLI Y et al. Synthèse et activités pharmacologiques des dérivés du pyrazole: une revue [J]. Molécules, 2018, 23(1):134.
[47]WANG J, WANG D B, SUI L L, Et al. Natural products — isoxazole hybrids: A review of Les développementsin medicinal chemistry [J]. Arabian Journal of Chemistry, 2024, 17(6):105794.
[48]DAI R, WEI X, LI T et al. Synthèse et activité antitumorale des dérivés du panaxadiol pyrazole et de l’isooxazole [J]. Chimie &; Biodiversité, 2023, 20 (8): e202300507.
[49]HOU W, DAI W, HUANG H et al. Activité pharmacologique et mécanisme des pyrazines [J]. European Journal of Medicinal Chemistry, 2023, 258:115544.
[50] rachid H U, U,U, Les MARTINES M A U, DUARTE DUARTE A P, et al. La recherche developments in the Synthèses, activités anti-inflammatoires et relations structure-activité des pyrimidines [J]. RSC Advance. 2021, 11(11): 6060-6098.
[51]WANG S, ZHANG J, ZHANG J, et al. synthèse and biological Évaluation des projets of hétérocyclique Dérivés de protopanaxadiol fusionnés en anneau en tant qu’agents antiostéoporose puissants [J]. Journal of Medicinal Chemistry, 2023, 66 (17): 11965-11984.
[52]YANG K, YANG Z, Yu G et al. Les nanomédicaments polypromédicaments: un paradigme émergent pour le traitement du cancer [J]. Advanced Materials, 2022, 34 (6): e2107434.
[53]LU H, Le CEN J, Mon - sun Y, et al. Evaluation of the anti-inflammatory La douleur effet of Particules d’o-carboxyméthyl chitosan conjuguées au ginsenoside [J]. Polymères, 2023, 15 (19): 4011.
[54]MATHIYALAGAN R, WANG C, KIM Y, et al. Préparation de polyéthylène glycol-ginsénoside Rh1 Et les conjugués Rh2 et leur efficacité contre le cancer du poumon et l’inflammation [J]. Molécules, 2019, 24 (23): 4367.
[55]BATHEJA S, GUPTA S, TEJAVATH K K, et al. Conjugués à base de TPP: ligands potentiels de ciblage [J]. Drug Discovery Today. 2024, 29(6):103983.
[56]MA L, WANG X, LI W,et al.Rational Conception, synthèse et évaluation biologique du triphénylphosphonium-ginsénoside conjugués as Ciblage des mitochondries anti-cancer agents [J]. Bioorganic Chemistry, 2020, 103: 104150.
[57]ZHANG H R, YE AQ, ZHANG Y W, et al. Progrès de la recherche sur la dérivatisation du ginsénoside et ses activités biologiques [J]. Chinese Traditional and Herbal Drugs, 2022, 53(14): 4554-4567.
[58]YUAN S Z, WANG B, Zhou X, et al. Progrès de la recherche sur la biotransformation des ginsénosides rares [J]. Science and Technology of Food Industry, 2023, 44(12): 480-489.
[59]RRN S, LIU R, WANG Y et al. Synthèse et évaluation biologique des analogues K du composé ginsénoside en tant que nouvelle classe d’agents anti-asthmatiques [J]. Biobiologique & Lettres de chimie médicinale, 2019, 29 (1): 51-55.
[60]REN G, LV W, DING Y et al. Ginseng saponin métabolite 20(S)-protopanaxadiol soulage la fibrose pulmonaire par des voies de signalisation multi-cibles [J]. Journal of Ginseng Research, 2023, 47 (4): 543-551.
[61]ZHANG H, ZHANG L, YANG C et al. Effet préventif des saponines de type protopanaxadiol saponines et protopanaxatriol Les saponines on myélosuppression souris induced by cyclophosphamide [J]. frontières Dans Pharmacology, 2022, 13: 845034.
[62]KIM A, PARK SM, KIM NS, et al. ginsénosideRc, un composant actif de panax ginseng, soulage l’atrophie musculaire induite par le stress oxydatif par l’amélioration de la biogenèse mitochondriale [J]. Antioxydants, 2023, 12 (8).
[63]LI S, LI JJ, ZHAO YY et al. Intégration supramoléculaire de nanomatériau multifonctionnel par azocalixarène décoré de mannose avec ginsenoside Rb1 Pour la thérapie synergique de la polyarthrite rhumatoïde [J]. ACS Nano, 2023, 17 (24): 25468-25482.
[64]LEE H, KONG G, TRAN Q et coll. Relation entre ginsénoside Rg3 Et le syndrome métabolique [J]. Frontières en pharmacologie, 2020, 11: 130
[65] fr Y Y, LIU Q P, AN P, et al. Ginsenoside Rd: A prometteur natural neuroprotection agent [J]. Phytomedicine, 2022, 95: 153883.
-
Précédent précédent
Quelle est la méthode d’extraction du ginsénoside?
-
Suivant:
Qu’est-ce que le ginsénoside Rh2 et son dérivé?




